
Published 27 November 2018 by Judith M. Reichel
Nobel Prize in Chemistry 2018 – The Evolutionary Tale of Enzymes and Antibodies
This blog post is part of a series of articles on the scientific research that led to this year’s Nobel Prizes. The official Nobel Prize Award Ceremony will take place in Stockholm on 10 December 2018.
Evolution and natural selection – those are the driving forces that turned us from the simple single-celled organisms into the complex, trillion of cells containing beings that we are today. But how could anyone ever ‘harvest’ the ‘power of evolution’? This year’s Nobel Laureates in Chemistry did just that.
The award went to three highly distinguished scientists – half of it was shared by George P. Smith, Professor Emeritus at the University of Missouri-Colombia, and Sir Gregory P. Winter, Professor at the University of Cambridge, UK. The other half was awarded to Frances H. Arnold, biochemist at the California Institute of Technology (Caltech). According to the Nobel Foundation, Frances Arnold received the prize “for the directed evolution of enzymes”. George P. Smith and Sir Gregory Winter were lauded “for the phage display of peptides and antibodies”.
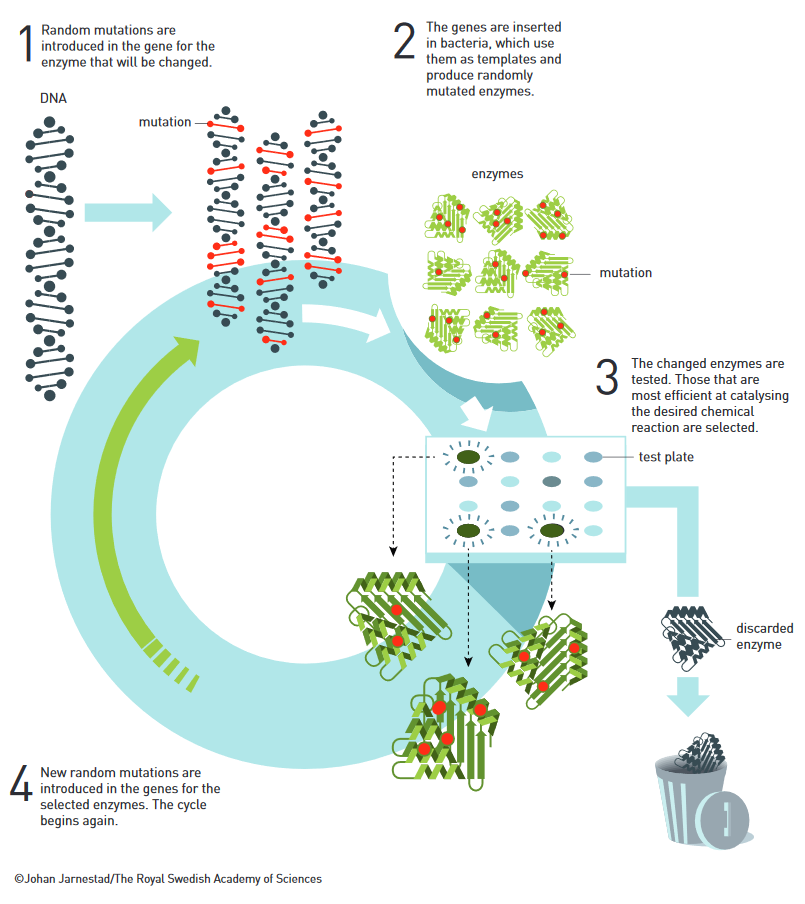
It was in the early 1990s at Caltech where Frances Arnold, born in 1956 in Pittsburgh, first developed what she was now awarded the Nobel Prize for: a method called ‘directed evolution’. This method is used to generate proteins that are not found in nature. Proteins are the building blocks of life. And enzymes, the catalysts involved in pretty much every biochemical reaction, are made up of proteins. Thus, the function of an enzyme and therefore of a biochemical reaction depends on the proteins involved. The structure and function of proteins, in turn, depends on the underlying DNA sequence. In order to generate a specific protein or enzyme function, Arnold induces DNA mutations within a specific DNA sequence of a protein with a function related to her target product. That altered sequence is then introduced back into a host organism or cell culture, where the sequence is translated into a new protein with an altered function. These altered functions are then tested, and the entire process is repeated over and over again until the protein and thereby the enzyme with the desired characteristics and functions is generated. What makes her method stand out is the fact that she is neither trying to design a new enzyme DNA sequence or protein structure from scratch, nor is she waiting for random mutations to introduce alterations that might or might not be useful (as is the case in evolution). Rather, by specifically inducing mutations at areas of interest, she is harvesting the random power of evolution for her benefit. Furthermore, by using rapidly proliferating bacteria as hosts for her newly synthesised DNA sequences, she is also able to quickly assess whether the proteins do in fact exhibit the desired functions – without needing to fully understand the complex co-dependence of protein-structure and its function. Since the beginning of the method in 1993, it has been refined several times and is now the gold-standard for developing new catalysts.
Another method to produce large amounts of peptides, proteins or antibodies is the so-called ‘phage display’. This method was first developed by George Smith, born in 1941 in Connecticut. The coating proteins of bacteriophages (or ‘phages’, in short), which are viruses that can infect bacteria, can be altered to specifically ‘display’ certain proteins. In 1985, Smith first introduced a protein sequence into a phage and the respective protein was subsequently ‘displayed’ on its surface. Such proteins on the phages are binding sites for peptides or other proteins. If such peptides or proteins bind to them, their binding dynamics and protein-protein interactions can be studied. Moreover, one phage can display several different binding sites. Thus, large libraries of proteins can be screened and selected for their functions and interactions, a process somewhat analogous to natural selection, where the ‘fittest’ version with the most appropriate functions survive or are ‘selected’.
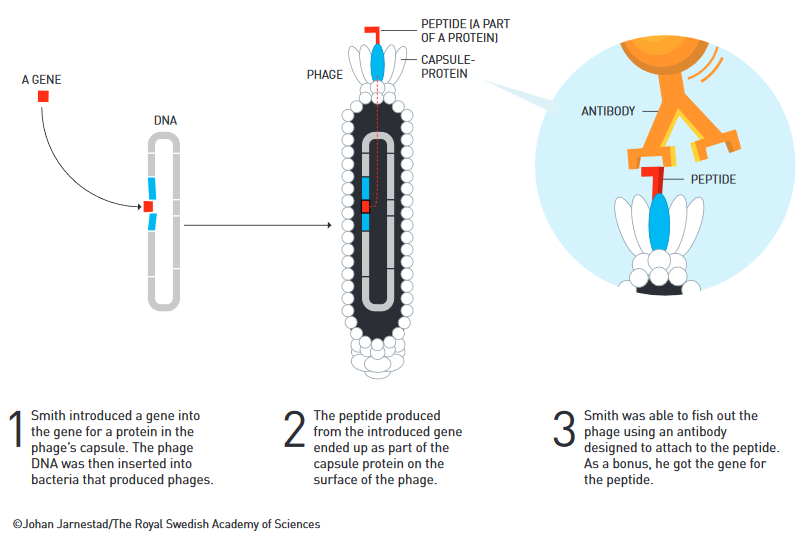
Finally, if the DNA sequence of a protein is known, this can be introduced onto the surface of the phage. If a peptide then binds to this surface protein, that specific binding site can be isolated and its genetic code be sequenced; this way a new antibody can be identified and subsequently generated in the lab.
Last but not least, Gregory P. Winter, born in 1951 in the Leicester, UK, developed this intricate method of phage display even further to produce new pharmaceuticals. Because of his improvements, many antibodies intended for human use no longer needed to be synthesised in mice or other host species, thereby minimising cross- and allergic reaction. Winter was so successful with his new antibody developing tool that he founded ‘Cambridge Antibody Technology’ in 1989 – one of the first biotech companies involved in antibody engineering. The first pharmaceutical antibody developed with this method was approved in 2002. It is an antibody to TNF alpha, a part of the inflammatory response, and is used to treat rheumatoid arthritis, psoriasis and inflammatory bowel diseases. It was the world’s top selling pharmaceutical in 2017 with sales over USD 18bn.
However, phage display as used by Winter is not only useful for broad diseases that affect all patients in a similar way. It has also become a very promising approach to treat (metastatic) cancers: Phage display is used to create and select synthetic antibodies that target the surface proteins of the tumour. These are then made into synthetic receptors for T-Cells collected from the patient, another essential part of the immune system. Thus, the patient’s own immune system is then able to recognise and fight the cancer cells, a process particularly useful for treating metastatic cancers.
Additional information: A new series of Mini Lectures concerning DNA and its replication and modification can be watched in our mediatheque.